Microscopic Trends Revealed on Electron-Hole Separation in Dye-sensitized Solar Cells
Date:10-06-2010 Print
By Yang Jiao and Sheng Meng
Dye-sensitized solar cells (DSSCs) are believed to be one of the most attractive renewable and low-cost energy solutions, which may replace fossil fuels in the 21st century. A DSSC benefit from its imitation of natural photosynthesis in that it separates sunlight absorption--which requires a large space--from electron collection processes which need highly pure materials and being most efficient on a small lengthscale. The biggest challenge to develop DSSCs is to realize both functions in the same system and to improve efficiency on both sides.By combining dye sensitizers with oxide semiconductor nanoparticles, DSSCs resolve this conflict. Visible light absorption efficiency is improved by >1000 times on nanoparticles compared to that of single-crystal surfaces, due to high surface/volume ratio of the former. Nevertheless, electron-hole separation dynamics at the dye/semiconductor interface, especially on a microscopic scale, remains elusive.
Professor Sheng Meng from Institute of Physics, Chinese Academy of Sciences/Beijing National Laboratory for Condensed Matter Physics, attacked this problem in collaboration with Prof. E. Kaxiras of Swiss Federal Institute of Technology at Lausanne. They examined the influence of various factors on the electron injection efficiency using state-of-the-art first-principles calculations within the framework of time-dependent density functional theory (TDDFT). On the basis of a previous work which illustrates an ultrafast electron-hole separation process at the anthocyanin/TiO2 interface [Nano Lett. 8, 3266 (2008)], they extended to demonstrate further that various factors including dye molecular size, binding geometry, and point defects on the TiO2 surface will greatly influence electron collection efficiency.
Experimentally it is observed that at various dye/TiO2 interface, the timescale of excited electron injection ranges from the shortest 3 fs (biisonicontinic acid, in vacuum) to 100ps (Ru-complex N719, triplet state injection, in devices). The huge time span suggests rich physical factors may play a role. Understanding this process will help us tune charge transfer timescale and improve sunlight-to-electricity conversion efficiency. For instance, electron injection will be 3.3 times slower with the addition of a CH2 group inserted between the dye molecule and the semiconductor, predicted from exponential decay of tunneling electron density when increasing separation distance in a non-adiabatic process. This was indeed observed in experiments using Re dyes (ReC1A-ReC3A). However, this pronounced time increase is not observed in experiments on Zn-porphyrins with one or four oligo(phenylethylny) bridges. From their TDDFT simulations, Meng and his coworkers found that among the three organic dyes they investigated longer molecules do involve a longer injection time, which is consistent with intuition. Furthermore, the time elongation is only 1.2 times (by inserting a (CH)2 group) or 1.3 times (inserting a thiophene group). This indicates that adiabatic processes play a major role in these cases, which also explains the Zn-porphyrin experiment.
They also found that dye adsorption configurations significantly affect electron injection. By comparing to measured spectroscopy, intact and dissociative dye adsorption are identified. The former is 30-50 fs slower than the latter. Different adsorption configurations of intact dyes result in injection time varies by three folds. The difference is mainly caused by the interface dipole moments. A positive dipole at the interface introduces an upshift of conduction band minimum (CBM), which will suppress excited electron transfer from the dye molecule to semiconductor conduction bands.
The semiconductor surface also imposes a fundamental influence on electron-hole dynamics. Dye adsorption on surface oxygen vacancies is very stable; it leads to a strong electronic coupling between the dye and the surface resulting in an electron injection time of ~50fs, 2-3 times faster than that on defect-free surfaces. But this improvement is at the cost of fast electron-hole recombination, which will reduce device efficiency. These simulations could explain well the two injection times at 40 fs and 200 fs observed in experiments, which would correspond to adsorption on defects and on clean surfaces, respectively.
They also studied electron back transfer after the excited electron has been completely injected into the TiO2. All the research results indicate that electrons and holes are separated in space at a time ~200 fs, to assure DSSCs work well. This work provides helpful insights and guidelines as to tune the nanoscale and ultrafast processes, and to maximize the energy conversion efficiency of DSSCs. The work is published in Nano Letters 10, 1238 (2010).
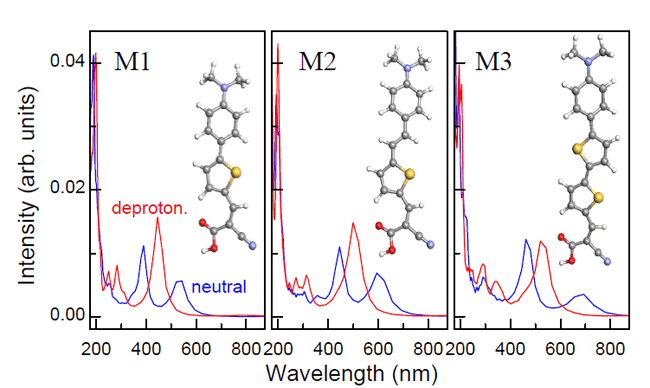 |
| Fig. 1. Absorption spectra of three organic dyes investigated. Blue lines: neutral, Red: deprotonated dyes. |
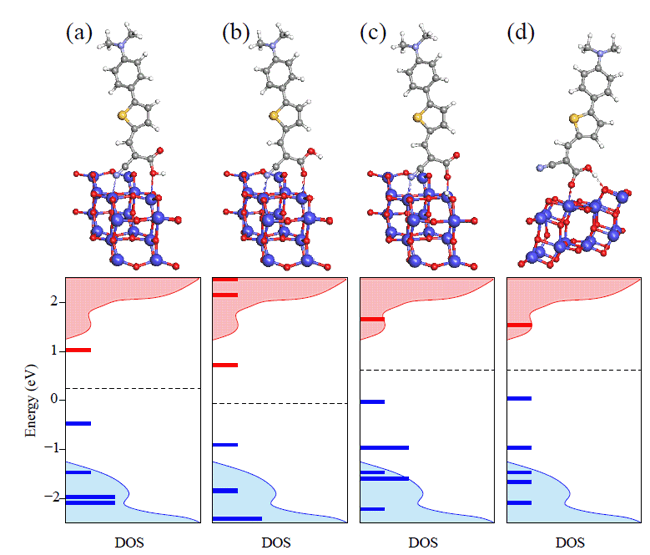 |
| Fig. 2 Adsorption configurations and corresponding electronic density of states for a dye on TiO2(101) surface. |
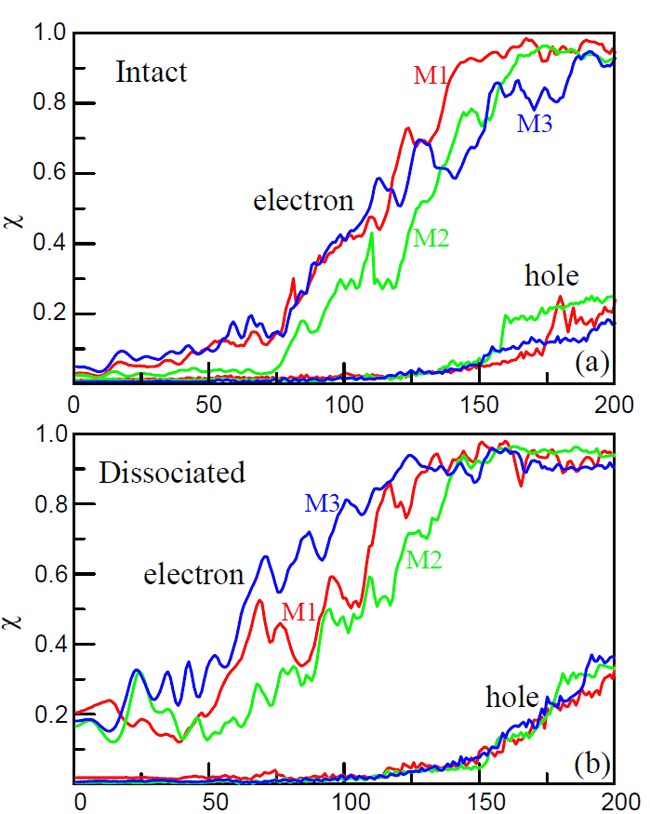 |
| Fig. 3 Electron-hole dynamics from TDDFT simulations. Vertical axis demonstrates charge injected into TiO2, horizontal axis denotes time simulated. |