Scientists reveal structural insights into NaV1.7 modulation by inhibitors
Date:27-11-2022 Print
Chronic pain is an extremely common disease that affects ~20% people of general population. Given the shortage of effective and non-addictive analgesics, new anti-pain drugs are eagerly awaited. Voltage-gated sodium channel NaV1.7 plays an essential role in the transmission of pain signals to the brain, many mutations in NaV1.7 had been directly linked to a variety of human pain disorders. Blockade of NaV1.7 can inhibit pain sensation, thus, NaV1.7 represents an attractive target for potential non-addictive analgesics.
Extensive efforts have been made in searching for NaV1.7 selective inhibitors by many programs, several candidate drugs have been undergoing clinical trials. However, NaV1.7 is a very challenging target for developing selective candidate drugs, partially owing to the high sequence similarity within the nine NaV channel isoforms. As a result, most of the candidate drugs failed due to the lack of selectivity or weak efficacy. Understanding the structural discriminations among NaV channel isoforms and the mechanism of how these inhibitors regulate NaV1.7 functions can support NaV1.7 related drug development, recently Zhang Jiangtao in Prof. JIANG Daohua's group from Institute of Physics, Chinese Academy of Sciences has reported cryo-EM structures of NaV1.7 in complex with three pore blockers, providing mechanistic insights into NaV1.7 modulation by pore blockers.
The researchers solved high-resolution cryo-EM structures of NaV1.7 complexed with three chemically distinct small molecule inhibitors XEN907, TCN-1752, and NaV1.7-IN2, respectively. The structure revealed the previously unresolved N-terminus domain (NTD) of NaV1.7 (Fig 1a-c), explaining that the conserved NTD is critical for NaV channel function. In addition, the structures confirmed that the central cavity of NaV1.7 accommodates multiple drug binding sites, which can directly block the channel (Fig 1b). Two of the three inhibitors also caused local conformational rearrangements of the S6 helix, that further affect the channel function.
The XEN907 caused a α-helix to the π-helix transition in S6IV of NaV1.7 (Fig 1d), which significantly slowed down the recovery of NaV1.7 from fast inactivation. The binding of TC-N1752 indirectly caused a local shift from the α-helix to the π-helix in S6II of NaV1.7, and shifted the S6II helix approximately 5 Å toward the activation gate, leading to a completely closed activation gate, and stabilizing NaV1.7 in the inactivated state (Fig 1e).
Further structural analysis revealed that the inhibitor binding sites located in the central cavity are highly conserved among the nine NaV channel isoforms (Fig 1f). Therefore, it is very challenging to achieve subtype-selective drugs binding in the central cavity of NaV channels. This study suggests that future efforts on searching for NaV1.7-selective inhibitors should focus on the regions that are relatively less conserved, such as the voltage-sensing domain (VSD).
This study entitled "Structural basis for NaV1.7 inhibition by pore blockers" was published on Nature Structural & Molecular Biology.
The study was supported by the National Science Foundation of China, Institute of Physics, and the Chinese Academy of Sciences.
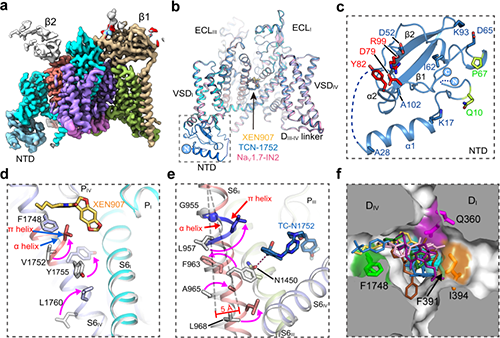
Fig. 1 a, Cryo-EM map of NaV1.7. b, Cartoon representation of NaV1.7 in complex with inhibitors. c, The structure of NTD. d, XEN907 binding site. e, TC-N1752 binding site. f, Superposition of inhibitors in the central cavity of NaV channel. (Image by Institute of Physics)
Contact:
Institute of Physics
JIANG Daohua
Email:jiangdh@iphy.ac.cn
Key words:
Voltage-gated sodium channel; NaV1.7; pore blockers; Cryo-EM; analgesics
Abstract:
Voltage-gated sodium channel NaV1.7 plays essential roles in pain and odour perception. NaV1.7 variants cause pain disorders. Accordingly, NaV1.7 has elicited extensive attention in developing new analgesics. Here we present cryo-EM structures of human NaV1.7/β1/β2 complexed with inhibitors XEN907, TC-N1752 and NaV1.7-IN2, elucidating specific binding sites and modulation mechanism for the pore-blockers. These inhibitors bind in the central cavity blocking ion permeation, but engage different parts of the cavity wall. XEN907 directly causes α- to π-helix transition of DIV-S6 helix, which tightens the fast inactivation gate. TC-N1752 induces π-helix transition of DII-S6 helix mediated by a conserved asparagine on DIII-S6, which closes the activation gate. NaV1.7-IN2 serves as a pore blocker without causing conformational change. Electrophysiological results demonstrate that XEN907 and TC-N1752 stabilize NaV1.7 in inactivated state and delay the recovery from inactivation. Our results provide structural framework for NaV1.7 modulation by pore-blockers, and important implications for developing subtype-selective analgesics.